3. User Guide¶
BLAST, HMMER, and Clustal are the main functions of the genomics-workspace. Each of these functions is implemented as a single app under Django.
In this section, we will go through details about how to configure each application.
In short, you need to configure database for BLAST and HMMER, but you don’t need to configure anything for Clustal.
Note
The page is for users that want to set up genomics-workspace by creating new admin user and confuguring in admin page. If you want to know how to use services provided by genomics-workspace, see these tutorials:
3.1. Getting started¶
To get started, set up the following:
- setup an admin account
- Use
python manage.py createsuperuser. - Follow the instructions shown on your terminal, then browse and login to the admin page of genomics-workspace. Usually, the admin page should be at
http://127.0.0.1:8000/admin/. - If you already have an admin account, use
python manage.py runserverand then browse and login to genomics-workspace.
- Use
- Create these directories if you don’t have them
- media/blast/db
- media/hmmer/db
- Create sequence sequence types in blast/sequence-type. We recommend creating these three:
- Peptide/Protein
- Nucleotide/Genome Assembly
- Nucleotide/Transcript
- Copy all fasta files to be formatted for blast to media/blast/db
- Copy protein fasta files to be formatted for hmmer to media/hmmer/db
3.2. BLAST Database Configuration¶
3.2.1. Manually creating a BLAST database¶
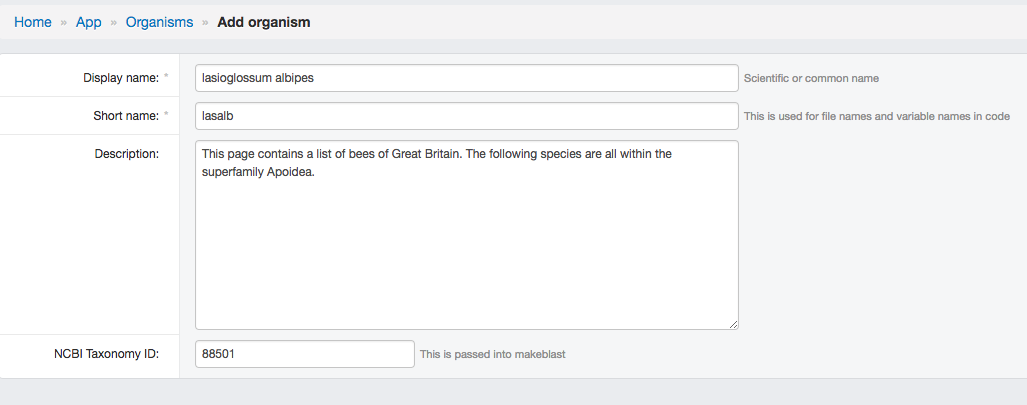
- Add Organism (click the Organism icon at sidebar and click Add organism):
- Display name should be scientific name.
- Short name are used by system as a abbreviation.
- Descriptions and NCBI taxa ID are automatically filled.
- Add Sequence
- Add BLAST DB
- Choose
Organsim - Choose
Type(Sequence type) - Type location of fasta file in
FASTA file path(It should be in<git-home>/media/blast/db/) - Type
Titlename. (showed in HMMER page) - Type
Descriptions. - Check
is shown, if not check, this database would show in HMMER page. - Save
- Choose
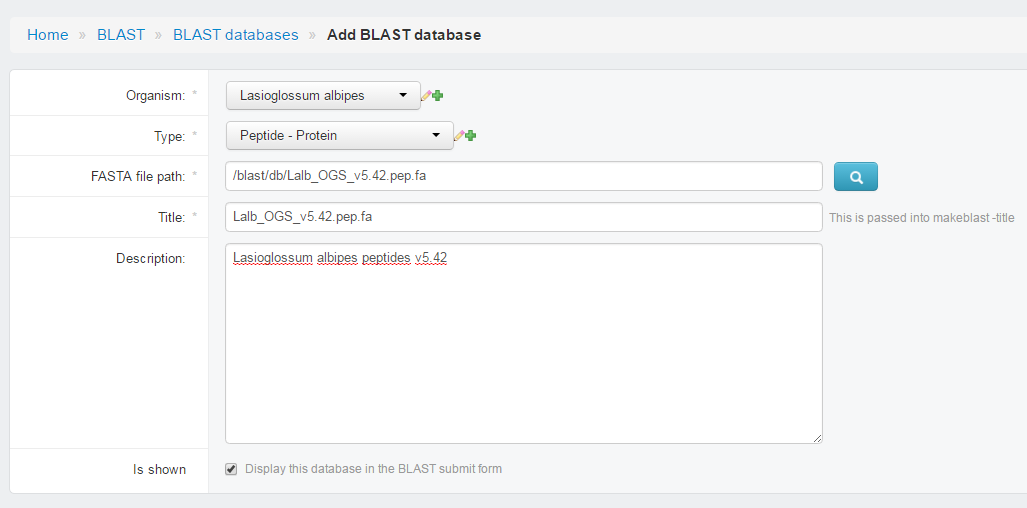
- Browse to
http://127.0.0.1:8000/blast/, you should able to see the page with dataset shown there.
3.2.2. Creating a BLAST database via command line¶
An admin user can add or remove data from the genomics-workspace database via the command line interface. Here, we describe how to use commands to interact with the database.
Note
the order of steps is important. Try to do these steps in order.
- To add organism
python manage.py addorganism [genus] [species](e.g python manage.py addorganism Apis mellifera)
- To add a fasta file to the Blast application
python manage.py addblast [genus] [species] -t [type] -f [path of fasta file] -d [description](e.g python manage.py addblast Apis mellifera -t nucleotide Genome Assembly -f media/blast/db/GCF_003254395.2_Amel_HAv3.1_genomic.fna -d Apis mellifera genome assembly, Amel_HAv3.1)- [type] here should be one of the sequence types you set up earlier, e.g. “peptide Protein”, “nucleotide Genome Assembly” or “nucleotide Transcript”
- [description] will be the Fasta file description in the web interface. If this argument is omitted, the program will use the Fasta file name. Example descriptions are “[genus] [species] genome assembly, [assembly name]”, “[genus] [species] [annotation name], peptides”, “[genus] [species] [annotation name], transcripts” or “[genus] [species] [annotation name], CDS”
- To make the blast database (via makeblastdb)
python manage.py blast_utility [path of fasta file] -m(e.g python manage.py blast_utility media/blast/db/GCF_003254395.2_Amel_HAv3.1_genomic.fna -m)
- To populate the genomics-workspace sequences table
python manage.py blast_utility [path of fasta file] -p(e.g python manage.py blast_utility media/blast/db/GCF_003254395.2_Amel_HAv3.1_genomic.fna -p)
- To show the blast database in the web interface (the blast database will not show by default)
python manage.py blast_shown [path of fasta file] -shown ‘true’(e.g python manage.py blast_shown media/blast/db/GCF_003254395.2_Amel_HAv3.1_genomic.fna -shown ‘true’)
3.3. HMMER Database Configuration¶
Like BLAST, HMMER databases must be configured then they could be searched.
Go to the django admin page and click Hmmer on the left menu bar. You need to create a HMMER db instance (Hmmer dbs) for each fasta file.
3.3.1. Manually creating a HMMER database¶
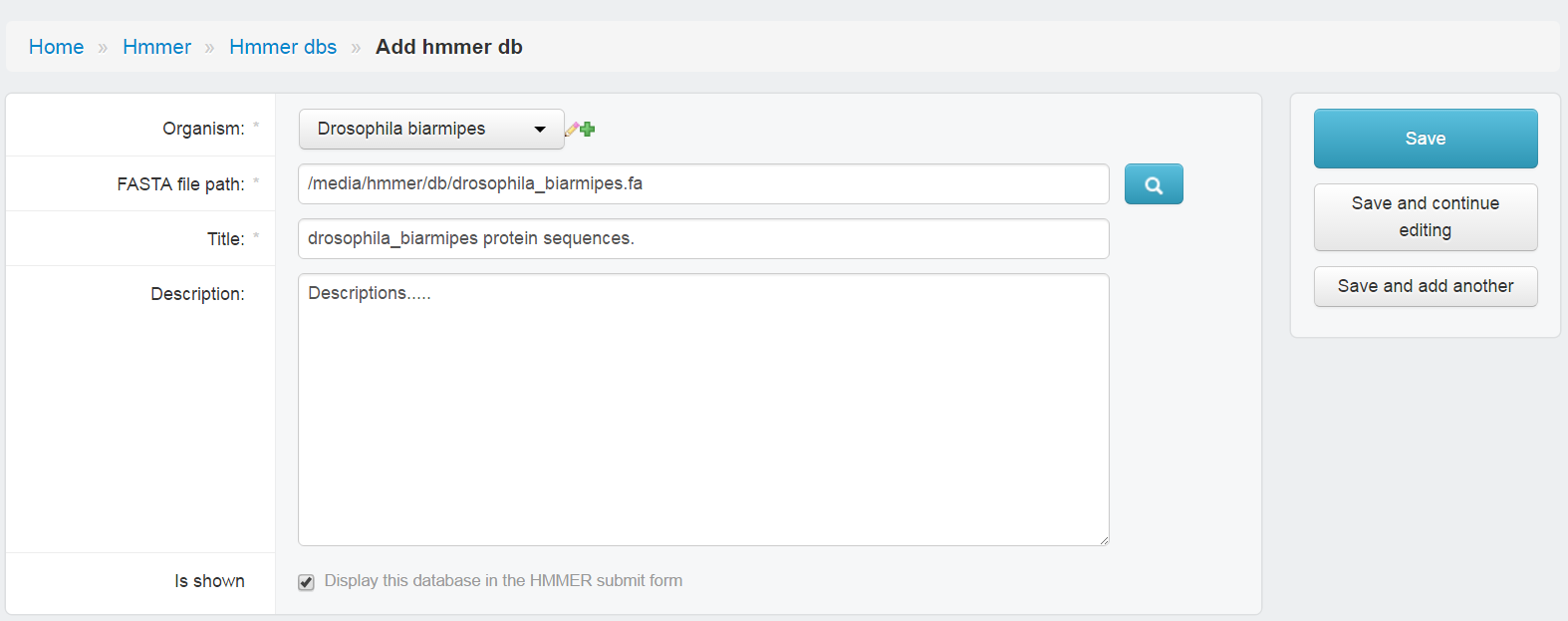
- Choose
Organsim - Type location of peptide fasta file in
FASTA file path - Type
Titlename. (showed in HMMER page) - Type
Descriptions. - Check
is shown, if not check, this database would show in HMMER page. - Save
3.3.2. Creating a HMMER database via command line¶
An admin user can add or remove data from the genomics-workspace database via the command line interface. Here, we describe how to use commands to interact with the database.
- To add organism (not necessary if the organism is already added)
python manage.py addorganism [genus] [species](e.g python manage.py addorganism Apis mellifera)
- To add hmmer
python manage.py addhmmer [genus] [species] -f [path of fasta file] -d [genus] [species] [annotation name], [sequence type](e.g python manage.py addhmmer Apis mellifera -f media/blast/db/GCF_003254395.2_Amel_HAv3.1_genomic.fna -d Apis mellifera Apis_mellifera_Annotation_Release_103, peptides)- [description] will be the Fasta file description in the web interface. If this argument is omitted, the program will use the Fasta file name. Example description: “[genus][ species] [annotation name], peptides”
3.4. Organism and Database deletion¶
Organism, BLAST and HMMER databases can be deleted after configuration via the command line interface. Here, we describe the commands for deleting them.
- To delete organism
python manage.py delete -o [genus] [species](e.g python manage.py delete -o Apis mellifera)
- To delete BLAST database
python manage.py delete -b [path of fasta file](e.g python manage.py delete -b media/blast/db/GCF_003254395.2_Amel_HAv3.1_genomic.fna)
- To delete HMMER database
python manage.py delete -h [path of fasta file](e.g python manage.py delete -h media/blast/db/GCF_003254395.2_Amel_HAv3.1_genomic.fna)